AlphaFold – changing the landscape of protein structure prediction


Structural biologists have an insatiable desire to discover the structure of proteins: for insight into how proteins work, and what that means for drug discovery. Traditionally, solving protein structures has been a slow, painstaking process, as most experimental techniques are simply too time consuming, or limited in the scope of which protein structures they can solve due to the associated costs - and of course, a little bit of luck is often needed, too! Over the decades, a panoply of theories and scientific approaches were developed to answer one of protein science’s biggest questions: how do the twenty standard amino acids fold to form the final protein structure? With recent advancements in computational approaches, namely DeepMind’s AlphaFold AI, we are one step closer to solving this challenge.
Solving a single protein structure can cost thousands of dollars, at a conservative minimum. These costs include labour, equipment time, reagent expenses and so on. To address this problem, a major focus of computational biology has been to discover a reliable method to predict protein structure from its sequence alone, without the expensive and time-consuming intermediate steps. To date, computational biologists have made steady progress, and have developed an array of structural prediction methods, such as now familiar tools like homology modelling, molecular dynamics simulations, as well as a litany of structure prediction algorithms (most notably those by the Baker Lab at the University of Washington). Community led efforts such as FoldIt and Folding@home have attempted to unravel the protein folding problem with a decentralised approach. These methods all seek to advance the solution to this core folding question of protein science, but so far, none have managed to produce a silver bullet due to insufficient accuracy, or high computational expense. However, this all changed with the arrival of an unlikely contender in the protein science realm: Google.
DeepMind, a British based subsidiary of Google – or rather, its holding parent company, Alphabet – has been a major contributor to AI projects in a broad array of fields beyond protein science. DeepMind’s project AlphaFold was a shock entry in the 2018 Critical Assessment of Techniques for Protein Structure Prediction (CASP13) competition. AlphaFold, DeepMind’s first attempt at a protein structure prediction program, proved superior to hundreds of prediction algorithms, including many that had been in the competition for years.
Still more surprisingly, their CASP14 attempt, using an updated AI named AlphaFold2 last year, achieved an unprecedented >90 out of 100 in two-thirds of sequences tested in the CASP’s global distance test. This attempt surpassed the generally accepted cut off for a high accuracy structure of 90, indicating a >90% accurate representation of the true molecule, rivalling the level of detail found in a traditional crystal structure. These consistently high scores stood head and shoulders above other groups’ results, representing a new benchmark for predictive algorithms, and was heralded as a transformative event in the field of protein science overall.

AlphaFold2 uses integrated neural networks to generate the final model, resulting in higher accuracy scores than the original AlphaFold, which had separately trained modules that often overfitted the final model with more secondary structure than necessary. In many cases, AlphaFold2 can predict structures with an accuracy comparable to the gold standard experimental techniques, such as X-ray crystallography or cryo-EM.
The benefits of having an algorithm that can predict protein structure from sequence alone cannot be understated – it will massively increase the speed of scientific discoveries in the treatment space. Furthermore, it will advance our understanding of the core science driving biological processes in the human body and beyond. In fact, since the public release of AlphaFold in mid-2021, every single protein biochemist colleague that I have worked with has talked about, asked for help with, or themselves used AlphaFold to move their projects forward. Protein structures, and the information they contain, informs almost every facet of work examining human and animal disease, from understanding how the human immune system works, to revealing the mechanisms of microbial infection. DeepMind itself has already published over 365,000 proteins predicted by their AlphaFold tool into an open-access database online, including predicted models of most proteins from the human and E. coli proteome, which will be a boon to biological researchers worldwide. DeepMind in partnership with EMBL plan to have millions of proteins modelled in only a few years time. Go look it up yourself – I’m sure you will find a protein there that you have struggled to solve for months, and it may be pretty close to what you thought it looked like!
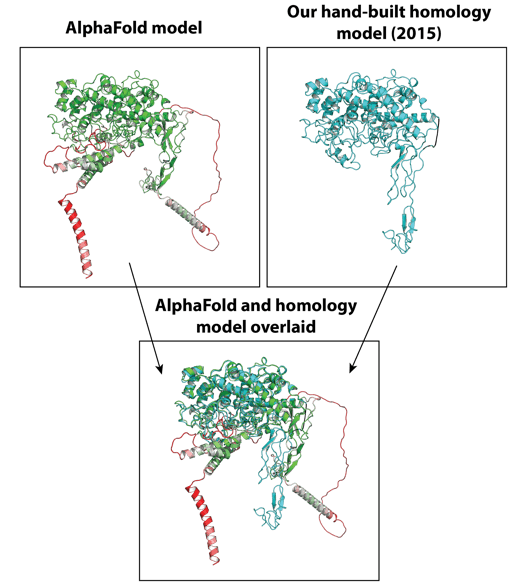
Researchers have long suspected that protein structure prediction would come to play a central role in therapeutic development and protein engineering. AlphaFold2 represents a powerful new tool for the protein engineering field for an endless list of reasons. Biological function can often be inferred from structure, and we can also anticipate rapid developments in ligand and candidate screening, prediction of drug-target interactions, rational design of engineered proteins, and improved drug target selection. AlphaFold can also improve our understanding of how intrinsically disordered proteins and intrinsically disordered regions within proteins may impact human disease and treatment, with these disordered regions suspected to be involved in approximately 30 percent of the human proteome.
A good structure or model increases the likelihood of a drug candidate that minimises side effects and maximises efficacy through superior design. With good modelling, we can predict mutations and create more stabilised, functional, and useful proteins for therapeutic or diagnostic applications. The Baker lab, the protein prediction pioneers, have developed designer proteins that could one day see the creation of a universal flu vaccine, or treat botulism, through this prediction method. The rise in AI and of neural networking techniques, such as those used by AlphaFold2, has pioneered the development of completely designed, de novo proteins. Drug design is an inherently time-consuming process, so expertise in computational approaches and how to apply them is critical for any lab to keep up with the fast pace of drug discovery. Of course, this need for speed has generated much interest in these systems in the biotechnology space – for example, AlphaFold was able to accurately predict and model SARS-CoV2 variants faster than other methods.
There were initial concerns that the Google-associated DeepMind would make AlphaFold a for profit, proprietary system. However, they have recently committed to making their developments open source – a triumph for the open and collaborative nature of science. The scientific community has already modified AlphaFold2 to generate models of complexes and oligomers, which AlphaFold2 had no native support for, using tricks involving residue indices and the inclusion of linkers. There was some criticism of AlphaFold for not being able to predict these complexes initially, the consequence of which pushed DeepMind to produce AlphaFold-Multimer, which has a more robust method of predicting multimeric and other complexes than other methods, showing how rapidly this group can adapt.
Needless to say, AlphaFold2 is a powerful tool for predicting protein structure. However, using AlphaFold requires computational expertise. To lower the barrier of entry for less computationally inclined scientists, enterprising developers created ColabFold, which has a simpler graphical user interface. However, this usability came at the price of reduced scope of functionality in some cases, and is less powerful compared to the command-line version of AlphaFold2, especially with more computationally intensive predictions. Over the decades, a multitude of computational tools have been developed in response to the growing demand from protein scientists. Computational biology is now its own area of expertise, as making the most of the technology available requires specialised knowledge of both computation and protein biology. This highlights the need for computational literacy amongst graduates in the field, or collaboration with teams who instead have this expertise.
Many scientists did not anticipate how quickly we have arrived at such an accurate algorithm as AlphaFold2, imagining that such a tool was years or even decades away, due to the computational expense of the pre-existing protein folding and prediction algorithms. It is perhaps no surprise that a project like might only come from a large software company such as Google – a company that has become increasingly dedicated and singularly focused on developing AI algorithms involving deep learning and neural networking, and has the financial backing that academic scientists often lack. As an added benefit of being part of such a large organisation, DeepMind had access to other Google technologies to create AlphaFold, such as Tensor Processing Units, a Google-developed dedicated hardware processing unit designed specifically for AI tasks. Despite this advantage in resources and finances, other projects in academia are quickly catching up to AlphaFold’s level of performance, such as RoseTTAFold.
AlphaFold is not a complete solution for the protein folding problem, as it cannot describe the steps that occur along the folding pathway – only the start and end points of sequence to structure (although some believe that the fold recognition problem has now been solved). However, DeepMind’s AlphaFold, AlphaFold2, and AlphaFold-multimer so rapidly surpassed what the scientific community thought achievable, that it will be only a matter of time before DeepMind (and other groups) begins solving protein folding mechanisms at an equally rapid pace. At this rate, they are the benchmark, though scientists around the world are swiftly closing the gap. Without a doubt, the ability to predict structure so easily and accurately is one of the major scientific advancements of this century. As we once mapped the first human genome, we now enter the realm of mapping out the diversity of structure that exists in the human proteome. It is an exciting time to be a protein biochemist!